Синдром Марфана
Синдром Марфана – это системное заболевание, передающееся по аутосомно-доминантному типу наследования, характеризующееся недоразвитием соединительнотканных волокон, вследствие возникновения структурных дефектов в коллагене. Наиболее часто при данной патологии поражаются органы зрения, опорно-двигательный аппарат, сердце и сосуды. Прогноз при этой болезни будет определяться выраженностью сопутствующих патологических изменений. Большинство пациентов не доживают до пятидесяти лет.
Впервые симптомы, характерные для синдрома Марфана, были описаны в 1875 году американскими врачами. Свое название данное заболевание получило в честь французского врача-педиатра А. Марфана, на протяжении нескольких лет изучавшего эту болезнь.
В настоящее время синдром Марфана встречается не часто. В среднем уровень его распространенности колеблется от 1 случая на 20 тысяч человек до 1 случая на 5 тысяч человек. При этом с ним одинаково часто сталкиваются как представители мужского, так и женского пола. Какой-либо расовой детерминированности также не прослеживается.

Суперфуды в косметике: сочные коктейли для здоровья кожи и волос
Симптомы
Симптомы синдрома Марфана отличаются своим многообразием.
Пациенты, страдающие от данного заболевания, как правило, имеют специфический внешний вид. Они отличаются высоким ростом при условии относительно небольшой длины туловища и чрезмерно длинных верхних и нижних конечностей. Помимо того, что руки и ноги больного человека непропорционально длинные, они еще и очень тонкие. Отмечаются удлинение и истончение пальцев, слабое развитие подкожно-жировой клетчатки, снижение мышечного тонуса.
Голова пациента имеет длинную и узкую форму. Нередко встречается аномалия развития верхней челюсти, представленная выгибанием твердого неба вверх с образованием высокого свода. В результате челюстно-лицевой деформации наблюдается неправильный прикус.
Суставы больного человека избыточно подвижны (гипермобильны), грудная клетка деформирована с выступанием вперед (килевидная форма) или западением во внутрь (воронкообразная форма) грудины. Кроме этого, характерны различные деформации со стороны позвоночного столба, например, его искривление в передне-задней плоскости.
Типичным признаком синдрома Марфана являются различные патологические изменения со стороны сердечно-сосудистой системы. Они могут быть представлены дефектами сосудистых стенок, например, аневризмами, пороками развития клапанов и перегородок сердца, например, пролапсом митрального клапана, расширением корня аорты, и так далее. В 2013 году ученые из Национального медицинского исследовательского центра сердечно-сосудистой хирургии им. А.Н. Бакулева опубликовали результаты работы, в которой было установлено, что расширение корня аорты встречается у 60% пациентов с синдромом Марфана, пролапс митрального клапана – у 91%.
Кроме этого, при данном заболевании зачастую поражаются органы зрения. С клинической точки зрения это может проявляться миопией различной степени выраженности, эктопией хрусталика, косоглазием, патологическими изменениями со стороны роговицы и так далее.
Помимо вышеперечисленного, синдром Марфана может сопровождаться поражением и других органов, например, нервной или дыхательной системы, кожи и многого другого.
Формы
Формы синдром Марфана могут быть следующие:
Стертая форма — слабо выраженные патологические изменения обнаруживаются в одной или двух системах организма.
Выраженная форме – могут отмечаться слабо выраженные симптомы со стороны трех и более систем, выраженные симптомы со стороны одной, двух и более систем.
Кроме этого, данное заболевание может иметь прогрессирующее или стабильное течение.
Причины
Причина синдрома Марфана – это генетическая мутация, передающаяся по аутосомно-доминантному типу наследования.
При этом ведущая роль в возникновении данной патологии отводится мутации в гене FBN1, отвечающем за выработку специфического гликопротеида – фибриллина. В результате нарушения синтеза фибриллина изменяется структура коллагеновых волокон, появляются проблемы с образованием волокнистых структур, соединительная ткань теряет свою прочность и упругость, не может выдерживать физиологические нагрузки.
Примерно в семидесяти пяти процентах случаев у человека с данным диагнозом выявляются родственники, страдающие от аналогичной проблемы. Значительно реже мутация в гене FBN1 является первичной. Замечено, что вероятность рождения ребенка с данной болезнью находится в прямой зависимости от возраста мужчины. Чем старше отец, тем выше риски.
Методы диагностики
Диагностика синдрома Марфана начинается с объективного осмотра, сбора анамнеза.
В настоящее время разработаны диагностические критерии данного заболевания:
Со стороны опорно-двигательного аппарата большими критериями считаются наличие килевидной или воронкообразной формы грудной клетки, искривление позвоночника во фронтальной плоскости более двадцати градусов, смещение вышележащего позвонка относительно нижележащего, плоскостопие, смещение медиальной стенки вертлужной впадины в сторону полости таза и так далее.
Со стороны сердечно-сосудистой системы к большим критериям относятся расширение корня аорты или расслоение ее восходящей части.
Со стороны органов зрения единственным большим критерием является эктопия хрусталика разной степени выраженности, выявляемая в 50-80% случаев.
Со стороны нервной системы единственный большой критерий – это эктазия твердой мозговой оболочки.
Все остальные патологические изменения со стороны внутренних органов принято относить к малым критериям. Постановка диагноза «синдром Марфана» основывается на наличии как минимум одного большого критерия со стороны двух систем и одного малого критерия со стороны третьей системы. Кроме этого, диагноз считается подтвержденным при наличии четырех и более больших диагностических критериев со стороны опорно-двигательной системы.
Из инструментальных методов исследования при данном заболевании назначаются:
- Рентгенография грудной клетки и тазобедренных суставов.
- Электрокардиография.
- Эхокардиография.
- Аортография.
- Компьютерная и магнитно-резонансная томография сердца и сосудов.
- Офтальмоскопия, а также многое другое.
Для окончательного подтверждения диагноза могут прибегать к помощи генетического исследования.
Лечение
Лечение синдрома Марфана направлено на то, чтобы остановить прогрессирование болезни и не допустить развития осложнений, в особенности, со стороны сердечно-сосудистой системы.
Из лекарственных препаратов для купирования симптомов, указывающих на поражение сердца и сосудов, могут назначаться бета-адреноблокаторы, ингибиторы ангиотензинпревращающего фермента, антагонисты кальция. Недостаточность сердечных клапанов, аневризма грудного отдела аорты диаметром пять и более сантиметров, расслоение аорты – все это является показанием к хирургическому вмешательству.
Особого внимания требуют беременные женщины с наличием данной патологии. В 2016 году ученые из Омского государственного медицинского университета опубликовали работу, по результатам которой было установлено, что при синдроме Марфана частота расслаивающей аневризмы аорты во время беременности составляет 4,5-6% случаев. При расслоении аорты в первом и втором триместре беременности необходимо провести экстренное кардиологическое хирургическое вмешательство, в третьем – сначала оперативное родоразрешение, а только затем протезирование аорты.
В остальном тактика лечения также будет определяться имеющимися патологическими изменениями. Так, например, при наличии катаракты требуется ее удаление при помощи хирургического или лазерного вмешательства, при деформации грудной клетки – нужна торакопластика.
Профилактика
Профилактика синдрома Марфана не разработана.
Какие вопросы следует задать врачу
Что такое синдром Марфана?
Из-за чего развивается данное заболевание?
Какие симптомы характерны для синдрома Марфана?
Как можно подтвердить диагноз?
Какова тактика лечения при выявлении синдрома Марфана?
Советы пациенту
Поскольку какие-либо способы предупредить развитие синдрома Марфана не разработаны, парам, собирающимся стать родителями, рекомендуется на этапе планирования беременности посетить врача-генетика для консультации и выявления возможных рисков.
Лечение синдрома Марфана и клинические рекомендации

Синдром Марфана – наследственная аномалия, для которой характерно системное поражение соединительной ткани. Заболевание проявляется изменениями в строении скелета, офтальмологическими патологиями, нарушениями функции сердца и дыхания. Предотвратить или вылечить болезнь невозможно. Но правильно подобранное лечение помогает спасти пациентов от многих осложнений и продлить им жизнь.
Причины синдрома Марфана
В развитии болезни «виновата» генетика. Синдром вызывается мутациями в гене FBN1, отвечающего за выработку фибриллина. Этот белок несет ответственность за прочность и эластичность соединительной ткани. Потеря соединительнотканными структурами своих главных свойств и становится причиной всех негативных изменений в организме.
Наследуется дефект от родителей, страдающих проявлениями синдрома. Шанс получить его у их детей составляет примерно 50%. Болезнь не передается через поколение: если у больных родителей родился здоровый ребенок, своим потомкам дефектный ген он не передаст.
Однако в 25% случаев синдром развивается у ребенка, рожденного от здоровых родителей. И конкретных факторов, вызывающих развитие синдрома Марфана, до сих пор не установлено.
Признаки мутации могут проявиться уже в первые месяцы жизни. Но нередко проявления болезни стерты и диагноз устанавливается уже во взрослом возрасте.
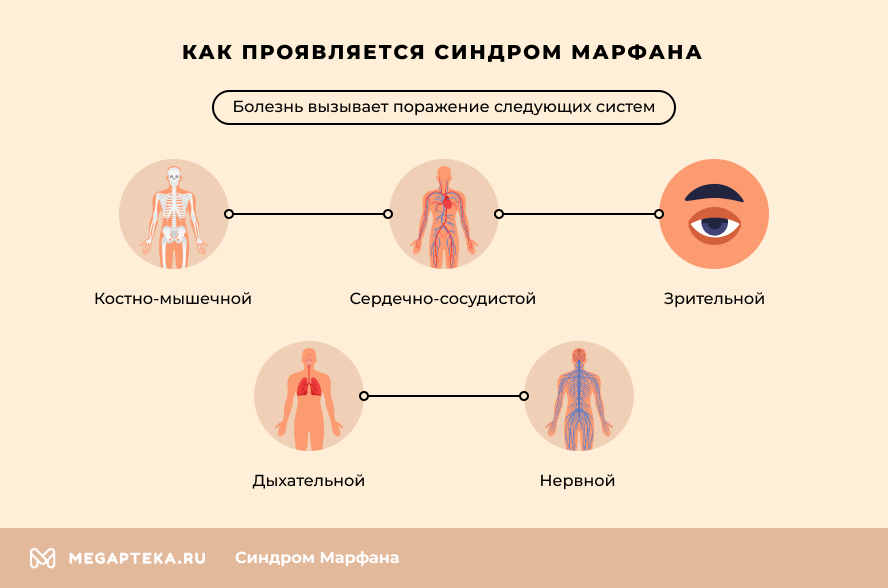
Как проявляется синдром Марфана
При стертой форме болезни поражаются 1-2 системы организма (например, скелетно-мышечная и/или органы зрения), а симптоматика проявляется незначительно. Несмотря на нарушения, такие пациенты живут практически нормальной жизнью. При выраженной форме поражаются 3 и более систем организма, либо наблюдаются значительные изменения в одной из систем.
Один и тот же генетический дефект проявляется по-разному – от незначительных изменений до тяжелых нарушений работы органов.
- Костно-мышечной системы – для пациентов характерен высокий рост, патологическая худоба, удлиненное лицо. Другие яркие симптомы – удлиненные и тонкие пальцы, нестандартная длина рук. Осанка в большинстве случаев нарушена, определяются сколиоз или кифоз. Нередко у пациентов наблюдается плоскостопие и повышенная подвижность суставов.
- Сердечно-сосудистой системы (ССС) – синдром вызывает нарушения сердечного ритма, недостаточность митрального клапана, различные пороки сердца. Особая опасность кроется в патологических изменениях в аорте. У большинства пациентов повышен риск расширения ее восходящей части и клапанного кольца, формирования аневризмы.
- Зрения – чаще всего наблюдается выраженная близорукость, подвывих хрусталика или изменение его положения. Также существует повышенный риск отслойки сетчатки. Нередко у больных уже в молодом возрасте развиваются глаукома и катаракта.
- Дыхательной системы – патологическое разрастание соединительной ткани в легких вызывает сужение бронхов и легочный фиброз. Нередко генетическая мутация приводит к развитию бронхиальной астмы.
- Нервной системы – в большинстве случаев нарушения мозговых структур отсутствуют. Но расширение соединительнотканной капсулы вокруг спинного мозга провоцирует нарушения движений ног, функции мочевого пузыря и кишечника. Высокий выброс адреналина часто вызывает повышенное нервное возбуждение и гиперактивность.
Также при синдроме Марфана могут наблюдаться опущение почек, матки, варикоз, патологии желудочно-кишечного тракта (ЖКТ).
Но физические недостатки природа компенсирует высоким уровнем умственных способностей. Нередко у таких пациентов выявляются неординарное мышление и повышенный интеллект.
Симптомы синдрома Марфана
Диагностика синдрома Марфана
- семейного анамнеза – уточнение данных о состоянии здоровья родителей;
- физикальной диагностики – наличие типичных признаков, в том числе, особенности антропометрических показателей;
- данных исследований – ЭКГ (электрокардиография), ЭхоКГ (эхокардиография) сердца, МРТ (магнитно-резонансная томография) мозга и позвоночника, рентгенологического, офтальмологического обследований;
- генетического обследования.
Лечение синдрома Марфана
Лекарственных методов терапии этой патологии, к сожалению, пока не разработано. Лечебные мероприятия зависят от конкретных проявлений болезни.
Если нарушения ССС выражены не сильно, назначают консервативную терапию. При значительном расширении восходящей части и расслоении аорты, пролапсе митрального клапана, пороках сердца применяют хирургические методы.
Коррекция зрения проводится с помощью очков и контактных линз. При необходимости выполняется хирургическое лечение глаукомы, катаракты, замена смещенного хрусталика искусственным.
Для поддержания опорно-двигательного аппарата применяется коллаген-нормализующая терапия, назначаются витаминно-минеральные комплексы. Выраженные скелетные нарушения корректируются хирургическим путем.
Клинические рекомендации исключают для пациентов с мутацией физические нагрузки, травмоопасные игры, высокую активность. Для поддержки позвоночника показано ношение корсета, с целью укрепления мышц – занятия ЛФК (лечебная физическая культура) и массаж.
Жизнь людей с этой генетической мутацией нельзя назвать простой. Но при ответственном отношении пациентов к своему здоровью уменьшить проявление болезни и предупредить серьезные осложнения вполне реально.
Синдром Марфана
Синдром Марфана — дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
МКБ-10
Общие сведения
Синдром Марфана — системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана — одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста — TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных — первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
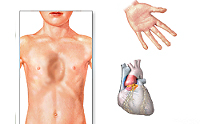
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана — миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца — коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ — обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов — выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Источник https://medaboutme.ru/zdorove/spravochnik/bolezni/sindrom_marfana/
Источник https://megapteka.ru/specials/sindrom-marfana-533
Источник https://www.krasotaimedicina.ru/diseases/children/marfan-syndrome